
Understanding Diluent & Reconstitution Studies in Oral and Parenteral Products
September 08, 2025
These studies bridge the gap between formulation science and clinical usability — ensuring that once the powder meets its diluent, the final product remains safe, potent, and stable through its intended shelf-life and usage period.

How to Perform an IID Search for Inactive Ingredients in US Drug Applications
September 08, 2025
This blog provides a step-by-step guide on how to perform an effective Inactive Ingredient Database (IID) search, particularly when preparing Module 3 (3.2.P.1) for US NDA/ANDA submissions. It emphasizes the importance of IID compliance to avoid Refuse-to-File (RTF) or Refuse-to-Receive (RTR) issues from the FDA. The blog also includes useful tips on filtering by route of administration, dosage form, and excipient grade, and highlights the importance of matching the intended use with previously approved quantities.
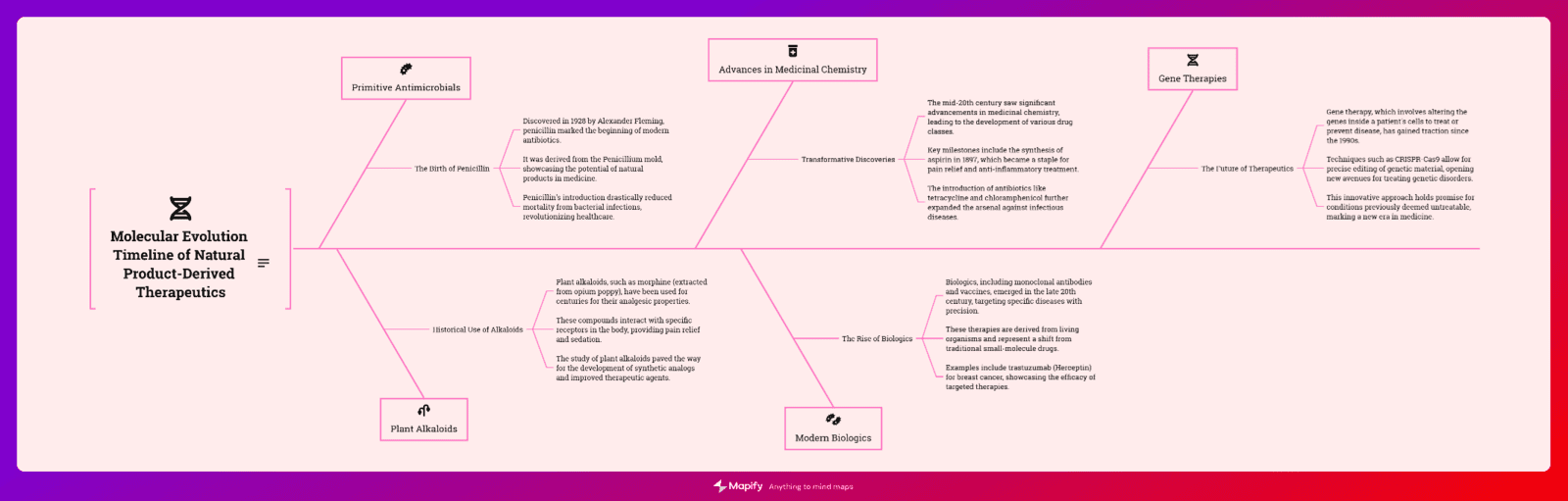
Structural Analogs and Derivatives of Natural Products in Modern Therapeutics
August 26, 2025
This paper presents an exploration of natural product derivatives in modern therapeutics
I. Introduction: From Nature to Laboratory
The journey from natural compounds - therapeutic innovations represent pharmacology's profound achievements.

Double Strand Break Repair (DSBR):
Mechanisms, Pathway Choice &
Clinical Significance
Mechanisms, Pathway Choice &
Clinical Significance
August 08, 2025
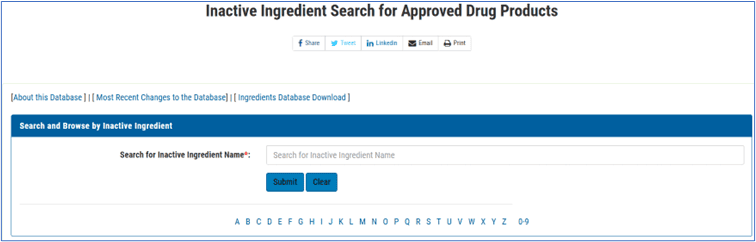
The Inactive Ingredient Database (IID) and Excipient Safety
1. The Inactive Ingredient Database (IID): Purpose and Utility
The Inactive Ingredient Database (IID) is a crucial resource for drug development, particularly for generic drugs.
August 05, 2025

US FDA Suitability Petitions - Updates
At ISAZI, we believe timely and precise regulatory intelligence is critical to informed decision-making in pharmaceutical development.
As part of our commitment to staying current and sharing value with the life sciences community, we are pleased to announce a new initiative: regular updates on Suitability Petitions (SPs) posted on Regulations.gov.
July 28, 2025

Regulatory Requirements for Elemental Iron Content in US ANDA Submissions (Module 3.2.P.1)
An ANDA must contain either a daily elemental iron calculation for products that contain iron or a statement that the amount of elemental iron ingested per day does not exceed 5 milligrams (mg), in accordance with 21 CFR 73.1200(c). A daily elemental iron calculation should be included in module 3.2.P.1 in addition to all other inactive ingredient justification data/information. If FDA does not receive either the calculation or aforementioned statement within 7 calendar days of notification of the omission(s) or product composition exceeds the elemental iron content more than 5 mg /day then FDA will RTR the ANDA.
July 28, 2025
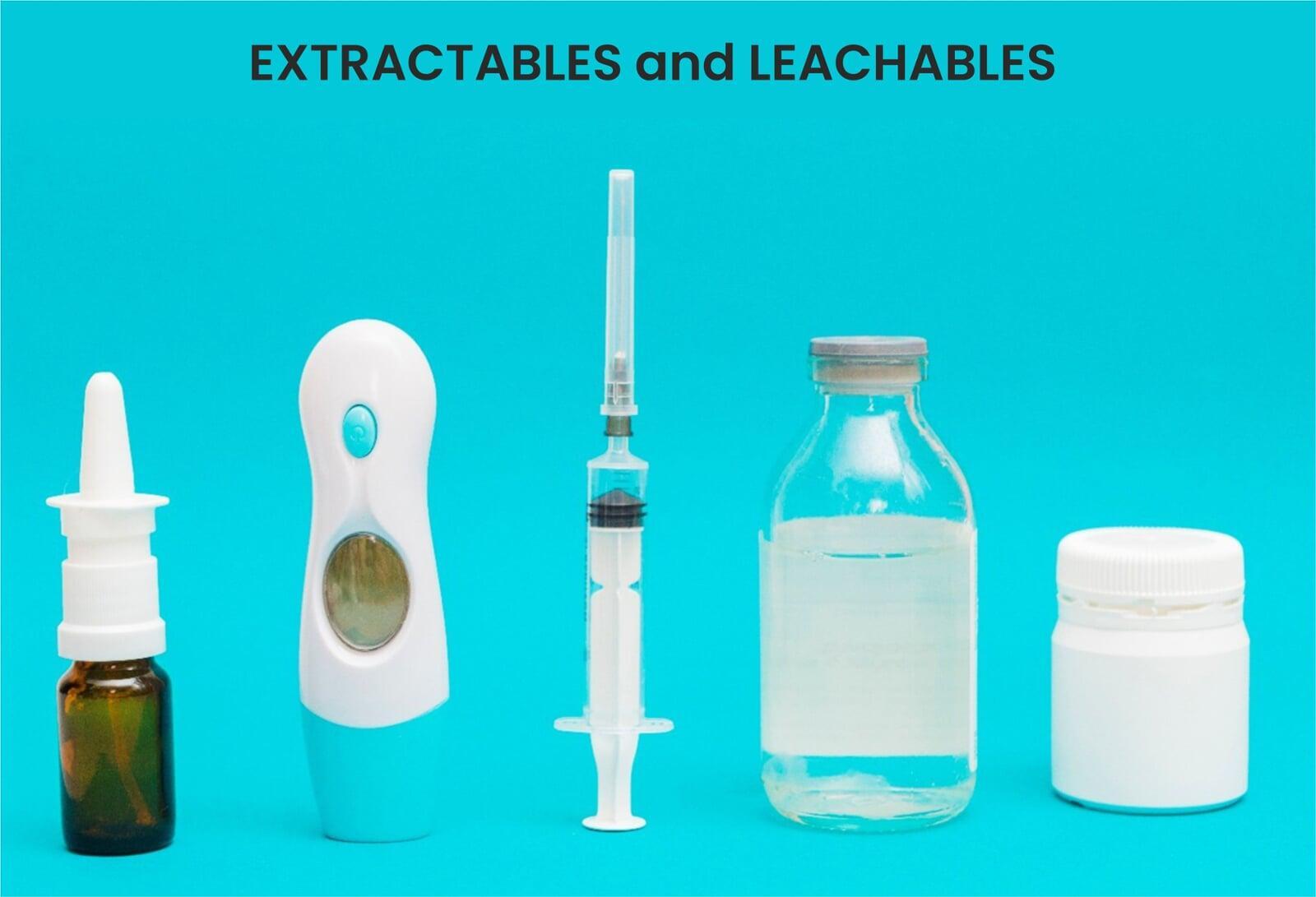
Demystifying Extractables and Leachables in the Pharmaceutical Industry: A Comprehensive Guide
In pharmaceutical product development, ensuring the safety, efficacy, and quality of the final product is of paramount importance. One critical but often overlooked area in this regard is the evaluation of extractables and leachables (E&L). These substances can originate from packaging materials, manufacturing components, or delivery systems and potentially contaminate drug products. Their presence, even in trace amounts, can lead to toxicity, loss of potency, or instability of the formulation.
July 26, 2025

CEP vs ASMF vs US DMF Type II: A Strategic Comparison
In the global pharmaceutical landscape, regulatory documentation for Active Pharmaceutical Ingredients (APIs) plays a pivotal role in ensuring product quality, safety, and compliance. While the US Drug Master File (DMF Type II) is widely recognized in the United States, the Certificate of Suitability (CEP) and Active Substance Master File (ASMF) are key submission formats in the European Union (EU). Understanding their differences is essential for crafting effective regulatory strategies across markets.
July 24, 2025

Specific aspect of the CC Requests Related to Inactive Ingredients
Generic drug manufacturers and related industry can submit written inquiries to the Office of Generic Drugs (OGD) that are referred to as controlled correspondence. These are requests for information on a specific element of generic drug development or certain postapproval submission requirements. FDA and industry agreed to timelines for the review of controlled correspondence under the reauthorization of the Generic Drug User Fee Amendments (GDUFA III).
July 22, 2025

Simplify Complex Generic – Things to Know About Pre ANDA Meetings (PDEV & PSUB)
To facilitate development of complex products that may be submitted in an ANDA, FDA and industry agreed to different types of meetings between applicants and FDA to discuss the proposed complex product and support submission of a high-quality, approvable ANDA, as well as to provide or continue to provide targeted, robust advice as applicants work to meet the standards for ANDA approval.
July 18, 2025

Force Degradation Study and Mass Balance
What is Force Degradation Study and Mass Balance:
A forced degradation study, also known as stress testing, is a process used in pharmaceutical development to evaluate the stability of drug substances and products by intentionally degrading them under harsh conditions beyond those of accelerated stability testing. This helps in identifying potential degradation pathways, developing stability-indicating analytical methods, and setting appropriate specifications for impurities.
July 15, 2025

Dissolution Discrimination: A Critical Tool in Drug Development and Regulatory Success
In the intricate world of pharmaceutical formulation, dissolution testing is more than just a quality control metric—it's a predictive tool for in vivo performance. Among its many facets, dissolution discrimination stands out as a cornerstone for ensuring product consistency, bioequivalence, and regulatory compliance.
July 10, 2025
Dissolution Discrimination: A Critical Tool in Drug Development and Regulatory Success
In the intricate world of pharmaceutical formulation, dissolution testing is more than just a quality control metric—it's a predictive tool for in vivo performance. Among its many facets, dissolution discrimination stands out as a cornerstone for ensuring product consistency, bioequivalence, and regulatory compliance.
July 10, 2025
Disclaimer:
The blogs on this page reflect the views of the author and should not be construed to represent any Agency’s or organization's views or policies. ISAZI is not liable for any damages, losses, or consequences that may arise from the use of this information or referencing it. The information is provided for academic interest and general information purpose only. Readers must ensure compliance with all applicable regulations and guidelines.
You can edit text on your website by double clicking on a text box on your website. Alternatively, when you select a text box a settings menu will appear. your website by double clicking on a text box on your website. Alternatively, when you select a text box